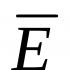Експоненційний закон надійності у кінетиці інактивації ферментів. Загальні властивості ферментів Вплив pH середовища на активність амілази слини
Релаксаційні методи засновані на тому принципі, що при швидкому зовнішньому впливі на систему (зміна температури, тиску, ін.) час, який потрібний системі для досягнення нової рівноваги (або стаціонарного стану), залежить від швидкості хімічної реакції (і іноді від швидкості дифузії реагентів .
Розглянемо найпростішу реакцію комплексоутворення активного центру ферменту з лігандом
Спочатку система знаходиться в рівновазі, яка характеризується константою рівноваги K 0 =K(T 0) і відповідно рівноважними концентраціями  ,
, ,
, . Припустимо, що у системі різко змінюється температура T->T 0 +T. Це призводить до зміни константи рівноваги K->K 0 +K, що визначається ставленням
. Припустимо, що у системі різко змінюється температура T->T 0 +T. Це призводить до зміни константи рівноваги K->K 0 +K, що визначається ставленням
 (2.50)
(2.50)
де H- Стандартна зміна ентальпії. Після цього система перетворюється на новий рівноважний стан:
 (2.51)
(2.51)
 (2.52)
(2.52)
Рівняння (2.51) нелінійне. Припустимо, що відхилення від рівноваги невелике і тоді
і рівняння (2.51) перетворюється на лінійне диференціальне рівняння:
Розв'язання цього диференціального рівняння:
Величина
 (2.54)
(2.54)
називається часом релаксації.
2.5. Вплив температури та pH на швидкість ферментативних реакцій
Вплив цих факторів на швидкість елементарної хімічної реакції було розглянуто у гл.1. Особливість у тому, що ферментативні реакції є складними багатостадійними реакціями (що з багатьох елементарних реакцій). Крім того, стан молекул ферменту в розчині характеризується набором конформерів, що оборотно переходять один в одного. Конформаційні переходи молекули визначаються значною мірою температурою та рН розчину.
2.6. Інгібування ферментативних реакцій
Речовини, що пригнічують каталітичну активність ферментів, називаються інгібіторами . Розрізняють два основні класи інгібіторів – оборотні
 (2.55)
(2.55)
(Пестициди, зарин, зоман, аспірин та ін.)
і незворотні (інактиватори )
 (2.55)
(2.55)
(окис вуглецю, ціанід-іон, анальгін та ін.)
2.7. Інактивація ферментів
Біополімерні молекули (ферменти) є термодинамічно нестійкими і, як правило, з часом змінюють свою структуру та властивості. У більшості випадків процес інактивації може бути описаний як перехід між двома станами активного ферменту E aта неактивного E i :
 (2.56) Кінетика процесу описується відповідним диференціальним рівнянням
(2.56) Кінетика процесу описується відповідним диференціальним рівнянням
 (2.57)
(2.57)
і характеризується постійним часом
 (2.58)
(2.58)
Процес інактивації ферменту може мати різну фізико-хімічну природу. Найбільш загальним є теплова денатурація, що є істотною перебудовою макромолекули, зміна третинної і частково вторинної структури.
Для цілей інактивації можуть бути використані кавітаційний ультразвук, радіоактивне випромінювання та ін.
Зміна pH також може призвести до денатурації ферменту. При кожному значенні pH білок характеризується відповідним розподілом зарядів (іоногенні групи). При дуже низьких або дуже високих pH розподіл зарядів може суттєво поляризувати молекулу, призводити до появи ізомерів і незворотно конформувати її з руйнуванням структури активного центру. Наприклад:

Денатурацію ферменту викликають і денатуруючі агенти, що руйнують вторинну структуру білка (наприклад, сечовина), а також окислювальні процеси за участю кисню
При вивченні таких процесів важливу інформацію отримано релаксаційними методами. Як правило, конформаційні зміни супроводжуються зміною оточення ароматичних амінокислот – тирозину та триптофану (смуга поглинання випромінювання на 290 нм). Це проявляється у зміні спектрів поглинання та флуоресценції.
Оборотні конформаційні зміни зазвичай йдуть з часом 0.1-100 мс, а незворотні - 1-1000 хв.
приклад 1. Найпростіша кінетична схема інактивації з рівновагою конформерів:
 (2.59)
(2.59)
Кінетика процесу описується одним характеристичним часом
 (2.60)
(2.60)
приклад 2. Інактивації піддаються обидва конформери:
 (2.61)
(2.61)
 (2.62)
(2.62)
приклад 3. Більш загальний випадок для системи за участю nконформерів:
Часто ферменти в розчині утворюють димери, і димерної формі виявляються стабільніше. Тоді спостерігається дисоціативний механізм інактивації :
 (2.65)
(2.65)
Наступна схема відображає можливі механізми інактивації у процесі реакції (мономолекулярна інактивація вільної форми ферменту, мономолекулярна інактивація фермен-субстратного комплексу, бімолекулярна інактивація ферменту субстратом, бімолекулярна інактивація ферменту продуктом):
 (2.66)
(2.66)
Дискримінація механізмів інактивації та визначення кінетичних характеристик реакції зазвичай проводиться кількома методами:
аналізуючи залежність виходу препарату від концентрації ферменту;
встановлюючи зв'язок ступеня конверсії субстрату зі ступенем інактивованості ферменту;
проведення реакції при низьких ступенях конверсії субстрату та низьких концентраціях ферменту;
проведення реакції при великих концентраціях ферменту;
перединкубація ферменту з компонентами реакції;
використання інтегральних рівнянь реакції.
Кількість ферменту, що у тканинах у будь-який час, визначається відносними швидкостями його синтезу і розпаду, і навіть концентраціями різноманітних інгібіторів і активаторів. Як правило, розпад ферментів та зниження їх кількості в середовищі відбуваються повільно. Інгібування та активація ферментів можуть здійснюватись досить швидко – протягом секунд.
Існує багато методів для визначення та вираження активності окремих ферментів. Це зумовлено різноманіттям ферментів, наявністю та використанням для визначення їх активності різних субстратів.
Міжнародна біохімічна спілка запропонувала таке визначення одиниці ферменту: « За одиницю будь-якого ферменту приймається та його кількість, яка каталізує перетворення одного мікромолю субстрату на хвилину за заданих стандартних умов ». Число мікромолей і дорівнюватиме числу стандартних одиниць . Міжнародна комісія запропонувала, якщо це можливо, проводити визначення активності ферментів при 30 °С і оптимальних для ферментативної активності значеннях рН і концентрації субстрату.
Загальні властивості ферментів витікають з їхньої білкової природи . Ферменти термолабільні, їх активність залежить від рН середовища та вологості , в якій вони діють, а також від впливу активаторів та інгібіторів .
При підвищенні температури до певних меж активність ферментів посилюється. При досягненні оптимальної для ферменту температури, його каталітична активність буває найбільш високою. Оптимальна температура для багатьох ферментів лежить найчастіше в межах від 40 до 50 °С (оптимальна для рослинних ферментів – 50 – 60 °З, а ферментів тваринного походження – 40 – 50 °З). Однак оптимальна температура не є постійною і залежить від багатьох причин, і зокрема від тривалості нагрівання. Чим триваліша дія ферменту, тим оптимальна температура має бути нижчою .
В інтервалі температур від 0 до 50 ° С при підвищенні або зниженні температури на кожні 10 ° С активність ферментів зростає або падає в 1,4 - 2 рази. При подальшому нагріванні активність ферментів знижується, та при 80 - 100 ° С ферменти зазвичай повністю втрачають каталітичні властивості у зв'язку з денатурацією білка .
Температура інактивації (втрати активності) у різних ферментів неоднакова. Так, інактивація ферменту амілази у розчині відбувається при 70 °С, сахарази – при 59, трипсину та пепсину – при 65 °С. У сухому стані ферменти можуть переносити нагрівання до вищих температур. Але за дуже високих температур інактивація ферментів настає миттєво. При пастеризації, стерилізації, бланшуванні та кип'ятінні ферменти руйнуються .
Після теплової інактивації деякі ферменти відновлюють свою каталітичну активність. Прикладом може бути пероксидаза, яка навіть при нагріванні протягом 60 с до 150 °С не повністю втрачає каталітичні властивості. Тому пероксидазу вважають найбільш термостабільним ферментом.
При температурах нижче 0 ° С каталітична діяльність ферментів різко знижується, але все ж таки зберігається навіть при заморожуванні продуктів.
Реакція середовища істотно впливає на каталітичну активність ферментів. Ферменти змінюють свою розчинність, осмотичний тиск, в'язкість та інші властивості під впливом середовища рН. Вважають, що зміна ферментативної активності залежно від рН середовища пов'язана із зміною іонізаціїферментів, субстрату або фермент-субстратного комплексу .
Ферменти проявляють оптимальну активність лише у певних, властивих їм межах рН. Так, пепсин, що виділяється у сильнокисле середовище шлунка, має оптимум активності при рН 1,5 та 2,5. У той же час протеази, що виділяються підшлунковою залозою у дванадцятипалу кишку, мають оптимальну активність у лужній зоні рН, а оптимум дії трипсину лежить у межах рН 8 – 9. При значенні рН вище або нижче оптимальної активність ферментів знижується .
Більшість ферментів бувають найбільш активними в нейтральних, слаболужних або слабокислих середовищах. У міру зсуву значення рН від оптимального в кисле або лужне середовище активність ферментів падає.
Активатори та інгібітори(паралізатори) ферментів можуть відповідно посилювати чи послаблювати і навіть припиняти їхню діяльність. Активаторами ферментів є іони металів: Na + , K + , Rb + , Mg 2+ , Ca 2+ , Cu 2+ , Fe 2+ та сполуки, що містять сульфгідрильні групи: SH, HCN, H 2 S . Наявність у розчині зазначених металів або сполук у певній концентрації сприяє прояву повної активності деяких ферментів.
Всі ферменти піддаються інгібуванню в результаті денатурації або руйнування ферментного білка.
Сутність дії інгібіторів у більшості випадків полягає в тому, що вони поєднуються з активними групами або активними центрами молекули ферментів. Розрізняють інгібітори загального та специфічного характеру . До загальним інгібіторам, які пригнічують дію всіх ферментів , відносять солі важких металів (свинцю, срібла, ртуті), трихлороцтову кислоту та танін . Часто гальмування або припинення дії ферментів під впливом важких металів носить оборотний характер, і якщо в середу додати речовини, що утворюють сполуки з цими металами, активність ферментів відновлюється.
Специфічні інгібіторидіють лише певні ферменти. Так, синильна кислота діє лише на окислювальні ферменти, що містять в активному центрі залізо чи мідь. Синільна кислота входить у з'єднання з металами, і фермент втрачає активність.
У живій клітині регулювання дії ферментів здійснюється не лише за допомогою специфічних активаторів та інгібіторів, але також шляхом зв'язування ферментів на різних колоїдних структурах протоплазми. Таке зв'язування ферментів призводить до втрати активності. Звільнення ферменту із сполуки знову відновлює його каталітичну активність.
Ферменти інактивуються при дуже високих тисках . Однак після зняття тиску ферменти відновлюють свою каталітичну активність.
Дія ферментів сильно уповільнюється у сухих продуктах, проте повністю не припиняється. Результати активності ферментів можуть виявлятися зміні якості продукту – його потемнінні, погіршенні аромату, смаку, консистенції тощо.
Швидкість більшості ферментальних реакцій пропорційна концентрації ферменту, принаймні, на ранніх стадіях. Поза початкових стадій швидкість ферментативних реакцій падає.
Фермент утворює з субстратом комплекс, який дисоціює на вільний фермент та кінцевий продукт реакції:
де Е – фермент; S – субстрат; ЕS – ферментно-субстратний комплекс; Р – кінцевий продукт.
Кількість субстрату дуже велика порівняно з кількістю ферменту, і тому концентрація субстрату сильно впливає швидкість ферментативних реакцій. Якщо субстрат міститься у значному надлишку, то кількість продукту, що утворюється, пропорційно часу. У міру зменшення концентрації субстрату кількість кінцевого продукту (Р), що утворюється в одиницю часу, знижується.
Про наявність у розчині ферменту судять з його дії. Так, про наявність амілази в слині можна судити за здатністю слини оцукрівати крохмаль, про наявність шлункового пепсину - за його здатністю розчиняти з достатньою швидкістю яєчний білок або фібрин.
Регулюючи активність ферментів створенням відповідної реакції середовища, можна керувати швидкістю каталізованих ними реакцій, а також діяльністю ферментів, що містяться в харчових продуктах, що дозволяє здійснювати заходи щодо зберігання зерна, картоплі, плодів та овочів, виробництва низки продуктів (вина, чаю тощо) .).
Номенклатура та класифікація ферментів
У початковий період розвитку вчення про ферменти їм давали назви без певної системи, за випадковими ознаками, за назвою субстрату або типу реакції, що каталізується. Так, фермент пепсин отримав назву від грецького слова «пепсис» – перетравлюю, папаїн – від соку рослини папайї, багатого на фермент. Траплялося, що окремі автори одному й тому ферменту давали різні назви.
У зв'язку з бурхливим розвитком науки про ферменти - ферментології в 1961 р. постійним комітетом з ферментів при Міжнародному біохімічному союзі була розроблена сучасна номенклатура та класифікація ферментів. Відповідно до цієї класифікації назва ферменту складалася з хімічної назви субстрату та назви тієї реакції, яка здійснювалася ферментом. До латинської назви кореня субстрату, на який діє фермент (сахароза – сахараза), або до назви процесу, що каталізується цим ферментом (гідроліз – гідролази), додавалася закінчення«аза». Поруч із новими назвами багатьом ферментів збереглися старі, міцно які у наукову літературу (пепсин, трипсин, папаїн та інших.).
За сучасною класифікацією всі ферменти ділять на шість класів: оксидоредуктази; трансферази; гідролази; ліази; ізомерази; лігази (синтетази) . Класифікація ферментів ґрунтується на характері їх дії.
Кожен клас поділяють на підкласи, а кожен підклас – на групи.
Оксидоредуктази
Це ферменти, що каталізують окисно-відновні реакції , які відбуваються у живих організмах. Реакції окислення речовин у організмах завжди супроводжуються реакціями відновлення. Оксидоредуктази ділять на 14 підкласів (Найбільший клас ферментів).
Окислення протікає як процес відібрання водню (електронів) від субстрату, а відновлення – як приєднання атомів водню (електронів) до акцептора. Цю реакцію схематично можна подати у такому вигляді:
АН 2 + В = А + ВН 2
де АН 2 - речовина, що віддає свій водень і називається донатором; В – речовина, яка забирає водень і називається акцептором.
Окислення можуть піддаватися різноманітні речовини – вуглеводи, жири, білки, амінокислоти, вітаміни та інших.
Роль оксидоредуктаз у живих тканинах виконують великі групи дегідрогеназ і оксидаз , які звуться залежно від окислюваного ними субстрату. Так, фермент, що дегідрує яблучну кислоту, називається малатдегідрогеназою, дегідруючий етиловий спирт - алкогольдегідрогеназою і т.д.
У класі оксидоредуктаз основне значення мають дегідрогенази, які здійснюють реакцію дегідрування. Усі дегідрогенази ділять на дві групи : анаеробні та аеробні, які називають оксидазами .
Анаеробні дегідрогеназиявляють собою специфічні ферменти, що каталізують відщеплення водню від певних хімічних речовин і які його інші ферментам – переносникам водню. Ці дегідрогенази є двокомпонентними ферментами, в яких кофермент легко відокремлюється від білкової частини. Як кофермент до складу анаеробних дегідрогеназ можуть входити дві речовини - нікотин-амід-аденін-нуклеотид ( НАД ) або нікотин-амід-аделінін-нуклеотид-фосфат ( НАДФ ). Обидві ці речовини мають виключно високу реакційну окислювально-відновну здатність.
Відомо дуже багато анаеробних дегідрогеназ, що каталізують окислення різних органічних сполук. Так, лактатдегідрогеназа каталізує реакцію окислення молочної кислоти до піровиноградної, ізоцитратдегідрогеназу – окислення ізолімонної кислоти до щавлево-бурштинової.
До групи аеробний дегідрогеназ (оксидаз)відносять ферменти, до складу яких як кофермент входить вітамін В 2 , (рибофлавін ), тому такі ферменти називають флавіновими . Флавінові ферменти здатні віднімати водень від речовини, що окислюється, і передавати його іншим сполукам або кисню повітря:
2Н2О2 → 2Н2О+О2.
Віднімаючи водень від речовини, що окислюється і передаючи її кисню повітря, оксидаза може при цьому утворювати воду або перекис водню (Н 2 Про або Н 2 Про 2). До цієї групи ферментів відносяться поліфенолоксидаза, аскорбінатоксидаза, глюкооксидаза.
Поліфенолоксидазаявляє собою аеробну дегідрогеназу, для якої акцептором водню є газоподібний кисень .
Вона діє на о-дифеноли, поліфеноли, дубильні речовини та тирозин. Поліфенолоксидаза широко поширена у грибах та вищих рослинах, особливо її багато в зеленому чайному листі. Дія поліфенолоксидази пояснюється потемнінням на розрізі м'якоті плодів і овочів, картоплі, а також потемнінням свіжого чайного листа при його скручуванні. Поліфенолоксидаза відіграє велику роль як проміжна ланка при диханні рослин.
Фермент пероксидаза поряд з поліфенолоксидазою та цитохромоксидазою бере активну участь у процесах дихання рослин та захисних реакціях рослин проти фітопатогенних мікроорганізмів рослин.
Активна група пероксидази містить залізо . За допомогою ферменту пероксидази за рахунок перекису водню та деяких інших органічних перекисів відбувається окислення органічних сполук. Пероксидаза утворює комплексну органічну сполуку, внаслідок чого перекис активується і набуває здатності діяти як акцептор водню:
Багато органічних сполук реагують з киснем повітря і утворюють перекиси. Особливо легко утворюються перекиси при окисленні киснем повітря сполук, що мають ненасичені зв'язки: каротиноїдів, ненасичених жирних кислот, деяких вуглеводнів.
Фермент каталаза каталізує процес розщеплення перекису водню на воду та кисень:
До складу молекули каталази, як і пероксидази, входить залізо . Головне призначення каталази в організмі полягає в тому, що вона руйнує шкідливий для клітин перекис водню, що утворюється в процесі дихання.
Фермент ліпоксигеназа каталізує утворення перекисів і гідроперекисів при окислювальному псуванні жирів.
Швидкість ферментативної реакції, іншими словами, активність ферменту визначається також присутністю серед активаторів і інгібіторів: перші підвищують швидкість реакції і іноді модифікують її, другі - гальмують реакцію. Серед хімічних сполук, які впливають активність ферментів, зустрічаються різноманітні речовини. Так, НС1 активує дію пепсину, жовчні кислоти – панкреатичної ліпази; деякі тканинні ферменти (оксидоредуктази, катепсини, аргіназа), рослинна протеїназа папаїн та ін. значною мірою активуються сполуками, що містять вільні SH-групи (глутатіон, цистеїн), а деякі також вітаміном С. Особливо часто як активатори служать (виступають) іони двовалентних та іноді одновалентних металів. Багато ферментів взагалі не активні за відсутності металів. Так, при видаленні цинку вугільна ангідраза практично позбавлена ферментативної активності; навіть при дії цього ферменту цинк може бути замінений ніяким іншим металом. Відомі ферменти, дія яких активується рядом металів, зокрема, енолаза (див. Вуглеводний обмін) активується Mg 2+ , Mn 2+ , К + . У табл. 18 наведено приклади участі металів у дії деяких ферментів.
| Таблиця 18. Метали активації деяких ферментів 1 (1 Зазвичай важко провести кордон між металоферментами (метал пов'язаний комплексно і незамінний) і ферментами, що активуються металами (останні лише прискорюють реакцію і легко дисоціюють).) | |||
| Фермент | Метал | Фермент | Метал |
| Цитохроми | Fe | Амілаза | Са |
| Каталаза | Fe | Ліпаза | Са |
| Пероксидаза | Fe | Карбоангідраза | Zn |
| Триптофаноксидаза | Fe | Лактатдегідрогеназа | Zn |
| Гомогентизиказа | Fe | Уриказа | Zn |
| Аскорбатоксидаза | Сі | Карбоксипептидаза | Zn |
| Тирозиназа | Сі | Пептидази | Mg |
| Фенолоксидаза | Сі | Фосфатази | Mg |
| Ксантиноксидаза | Мо | Фосфоглюкокіназа | Mg |
| Нітратредуктаза | Мо | Аргіназа | Mn |
| Альдегідоксидаза | Мо | Фосфоглюкомутаза | Mn |
| Пептидази | З | Холінестераза | Mn |
Щодо ролі металів в активуючій дії ферментів наявні дані свідчать про те, що в ряді випадків іони металів (2+, Mg2+, Zn2+, Fe2+) виконують функції простетичних груп ферментів. В інших випадках вони сприяють приєднанню субстрату до активного центру та утворення фермент-субстратного комплексу. Наприклад, іони Mg 2+ через негативно заряджену фосфатну групу забезпечують приєднання монофосфорних ефірів органічних речовин до активного центру фосфатаз, що каталізують гідроліз цих сполук. У ряді випадків метал з'єднується із субстратом, утворюючи справжній субстрат, на який діє фермент. Зокрема, іони Mg 2+ активують креатинфосфокіназу завдяки утворенню істинного субстрату та магнієвої солі АТФ. Нарешті, є експериментальні докази прямої участі металів (наприклад, іонів Са 2+ у молекулі амілази слини) у формуванні та стабілізації активного центру та всієї третинної структури молекули ферменту. Слід зазначити також нерідку роль металів як алостеричних модульаторів (див. рис. 59). Взаємодіючи з алостеричним центром, подібний метал (модулятор) сприяє утворенню найвигіднішої просторової конфігурації ферменту та активного фермент-субстратного комплексу.
Аніони при фізіологічних концентраціях зазвичай неефективні або мають невеликий активуючий вплив на ферменти. Виняток становлять пепсин, деякі оксидоредуктази, що активуються аніонами, а також слинна амілаза, що каталізує гідроліз крохмалю, активність якої підвищується іонами хлору, і аденілатциклаза, яка активується аніонами галогенів.
Інгібіторами прийнято називати речовини, що викликають часткове або повне гальмування реакцій, що каталізуються ферментами. Оскільки ферменти є білками, будь-які агенти, що викликають денатурацію білка (нагрівання, кислоти, луги, солі важких металів) призводять до інактивації ферменту. Проте таке інактивування щодо неспецифічне. Воно пов'язані з механізмом дії ферментів. Набагато велику групу становлять так звані специфічні інгібітори, які мають свою дію на один якийсь фермент або на групу родинних ферментів. Дослідження цих інгібіторів має важливе значення з низки причин.
По-перше, інгібітори можуть дати цінну інформацію про природу активного центру ферменту, а також його функціональні групи та хімічні зв'язки, що забезпечують утворення фермент-субстратного комплексу. Відомі речовини, що специфічно пов'язують ту чи іншу групу в молекулі ферменту, виключаючи її зі сфери хімічної реакції. Зокрема, йодацетат IСН 2 -СООН, його амід та етиловий ефір, парахлормеркурибензоат ClHg - З 6 Н 4 -СООН та інші реагенти порівняно легко вступають у хімічний зв'язок з деякими SH-групами ферментів. Якщо такі групи суттєві для акту каталізу, то додавання таких інгібіторів призводить до повної втрати активності ферменту:
R-SH + IСН 2 -СООН --> HI + R-S-СН 2 -СООН
Ряд інших ферментів (холінестераза, трипсин і хімотрипсин) сильно гальмується деякими фосфорорганічними сполуками, зокрема діізопропілфторфосфатом (ДФФ), внаслідок блокування ключової гідроксильної групи серину в активному центрі (див. вище).
По-друге, інгібітори знайшли широке застосування в ензимології при дослідженні природи множинних форм ферментів та ізоферментів, що відрізняються не стільки по електрофоретичній рухливості, скільки по відмінності реакцій на той самий інгібітор.
За допомогою інгібіторів, що вибірково вимикають окремі стадії багатоступінчастого метаболічного процесу, можуть бути точно встановлені послідовність хімічних реакцій і природа ферментів, що беруть участь. Зокрема, цим шляхом при застосуванні йодацетату, фториду та інших інгібіторів був розшифрований гліколітичний шлях окисно-відновних перетворень глюкози до молочної кислоти в м'язовій тканині (див. Обмін вуглеводів), що налічує 11 стадій за участю 11 ферментів і 10 проміжків.
На інгібуванні ферментів заснований механізм дії багатьох токсинів та отрут на організм. Так, відомо, що при отруєннях синильною кислотою смерть настає внаслідок повного гальмування дихальних ферментів (цитохромоксидази), особливо клітин мозку. Токсичне впливом геть організм людини і тварин деяких інсектицидів обумовлено гальмуванням активності холінестерази - ферменту, що грає першорядну роль діяльності нервової системи.
Раціональна хіміотерапія - усвідомлене застосування лікарських препаратів у медицині, має спиратися на точне знання механізму їхньої дії, на біосинтез ферментів чи роботу в організмі. Іноді метод лікування хвороб людини включає застосування виборчих інгібіторів. Так, інгібітор трипсину, хімотрипсину та калікреїну трасилол широко використовується при лікуванні гострого панкреатиту. Виборча інгібіторна дія на ферменти деяких природних та синтетичних сполук (так званих антиметаболітів) в даний час є основою для розробки ефективних методів синтезу хіміотерапевтичних препаратів. На цьому шляху відкриваються широкі можливості регулювання як синтезу ферментів, так і інтенсивності метаболізму.
Типи інгібування.Незважаючи на те, що механізм дії більшості інгібіторів не з'ясований, зазвичай розрізняють оборотне та незворотне інгібування. Якщо молекула інгібітора викликає стійкі зміни чи модифікацію функціональних груп ферменту, такий тип інгібування називається незворотним. Найчастіше, проте, має місце оборотне інгібування, яке піддається кількісному вивченню на основі рівняння Міхаеліса – Ментен. Оборотне інгібування своєю чергою поділяють на конкурентне і неконкурентне, залежно від цього, вдається чи вдається подолати гальмування ферментативної реакції шляхом збільшення концентрації субстрату. У другому випадку підвищення концентрації субстрату не змінює ступінь інгібування ферменту.
Конкурентне інгібування може бути викликане речовинами, що мають структуру, схожу на субстрат, але трохи від структури істинного субстрату. Класичним прикладом такого типу інгібування є гальмування активності сукцинатдегідрогенази малоновою кислотою. Цей фермент каталізує окислення шляхом дегідрування бурштинової кислоти у фумарову відповідно до схеми:
Якщо в середу додати малонову кислоту (інгібітор), то в силу структурної подібності її до справжнього субстрату янтарної кислоти (наявність двох таких же іонізованих карбоксильних груп) вона реагуватиме з активним центром з утворенням фермент-інгібіторного комплексу (див. схему), проте при цьому перенесення водню від малонату немає. Так як структури субстрату - бурштинової кислоти та інгібітора - малонату все ж таки дещо відрізняються, вони конкурують за зв'язування з активним центром, і ступінь гальмування визначатиметься співвідношенням концентрацій малонату та сукцинату, а не абсолютною концентрацією інгібітора. Цей тип інгібування іноді називають інгібуванням на кшталт метаболічного антагонізму (рис. 56).

У загальній формі реакція взаємодії інгібітора з ферментом може бути наступним рівнянням:
![]()
Комплекс, що утворився, званий фермент-інгібіторним комплексом (EI), на відміну від ES не розпадається з утворенням продуктів реакції. Константу дисоціації EI-комплексу або інгібіторну константу (K 1) можна, виходячи з теорії Міхаеліса - Ментен, визначити як відношення констант зворотної та прямої реакцій:
![]()
т. е. інгібіторна константа прямо пропорційна добутку концентрації ферменту та інгібітора і обернено пропорційна концентрації ЕІ-комплексу.
Метод конкурентного гальмування знайшов широке застосування у медичній практиці. Відомо, наприклад, що для лікування деяких інфекційних захворювань, що викликаються бактеріями, застосовують сульфаніламідні препарати. Виявилося, що ці препарати мають структурну подібність до параамінобензойної кислоти, яку бактеріальна клітина використовує для синтезу фолієвої кислоти, що є складовою ферментів бактерій. Завдяки цій структурній подібності сульфаніламід, наприклад, блокує дію ферменту шляхом витіснення параамінобензойної кислоти з комплексу з ферментом, що синтезує фолієву кислоту, що веде до гальмування росту бактерій.
Деякі аналоги вітаміну В 6 та фолієвої кислоти, зокрема дезоксипіридоксин та аміноптерин (див. Вітаміни), діють як конкурентні, так звані коферментні інгібітори (або антивітаміни), що гальмують багато біохімічних процесів в організмі.
Неконкурентне інгібування викликається речовинами, що не мають структурної подібності до субстратів і часто зв'язуються не з активним центром, а в іншому місці молекули ферменту. Ступінь гальмування у часто визначається тривалістю дії інгібітора на фермент. При цьому типі інгібування завдяки утворенню стабільного ковалентного зв'язку фермент часто піддається повній інактивації, і тоді гальмування стає незворотним. Прикладами неконкурентного інгібування (інактивації) є дія йодацетату, діізопропілфторфосфату, а також діетил-n-нітрофенілфосфату та синильної кислоти, що полягає у зв'язуванні та виключенні функціональних груп або іонів металів у молекулі ферменту.
Для з'ясування питання про тип інгібування користуються рівнянням Міхаеліса – Ментен, графіком Лайнуівера – Берка та іншими більш удосконаленими рівняннями, наприклад, рівнянням Еді – Хофсті:
v = - До m (0/[S]) + V mах
та відповідними графіками у прямолінійних координатах. У наведених нижче графіках, побудованих у координатах v і [S], а також у координатах 1/v і 1/[S], V - максимальна швидкість реакції, V 1 - максимальна швидкість у присутності інгібітора, К 1 - інгібіторна константа; решта символів були представлені вище.

Видно, що при конкурентному типі інгібування (рис. 57) інгібітор збільшує значення К m (на величину, що дорівнює різниці в довжині відрізків, що відсікаються від осі абсцис), не впливаючи на максимальну швидкість. Це означає, що з досить високої концентрації субстрату [S] інгібітор витісняється молекулами субстрату з комплексу EI. При неконкурентному інгібуванні (рис. 58) інгібітор знижує величину максимальної швидкості. Якщо при цьому величина К m не зменшується, то говорять про неконкурентне інгібуванні. Подібний тип інгібування має місце при утворенні неактивних, важкодисоціюючих комплексів EI та (або) EIS. Часто, однак, має місце змішаний тип інгібування (іноді частково званий неконкурентним типом), при якому зниження V mах поєднується зі збільшенням До m . Це означає, що комплекс EI зберігає часткову активність, тобто здатність до утворення проміжного потрійного комплексу EIS, в якому субстрат зазнає уповільненого каталітичного перетворення. У поодиноких випадках ступінь гальмування активності ферменту може збільшуватися з підвищенням концентрації субстрату; для цього типу гальмування був запропонований досить неточний безконкурентний термін (від англ. uncompetitive). Один з механізмів такого гальмування обумовлений можливістю з'єднання інгібітора з комплексом ES з утворенням неактивного або потрійного комплексу ESI, що повільно реагує.
Таким чином, при графічному аналізі швидкостей ферментативних реакцій як функції концентрацій субстрату може бути отримана цінна інформація з кінетики ферментативних реакцій, що висвітлює можливий механізм ферментативного каталізу.
Регуляція активності ферментів
Вище було зазначено, що однією з унікальних властивостей живих організмів є дивовижна здатність до збалансованості катаболічних (біодеградативних) та анаболічних (біосинтетичних) процесів. Хоча в клітинах одночасно відбуваються процеси синтезу, розпаду та взаємоперетворення сотень і тисяч різноманітних речовин, є безліч регуляторних механізмів, що забезпечують сталість внутрішнього середовища організму. Деякі з цих регуляторних механізмів, серед яких важлива роль належить механізмам регулювання активності ферментів, будуть розглянуті нижче.
Вплив закону дії мас.У каталізованої ферментом оборотної хімічної реакції, наприклад A+B C+D, концентрація компонентів реакції і відповідно напрям реакції регулюватимуться впливом закону дії мас. Воно, зокрема, може бути показано у оборотній реакції трансамінування, що каталізується аланінамінотрансферазою:
Аланін + α-Кетоглутарат Піруват + Глутамат.
Цей тип регуляції відіграє, очевидно, лише обмежену роль, оскільки в реальних умовах реакція зазвичай протікає в одному напрямку, оскільки продукти, що утворилися, можуть виявитися субстратами для дії інших ферментів і виводитися зі сфери реакції; у цих випадках встановлюється швидше стійкий (стаціонарний) стан, ніж справжня рівновага.
Зміна кількості ферменту.У бактерій добре вивчений феномен індукованого синтезу ферментів при вирощуванні їх на середовищі, де єдиним джерелом вуглецю та енергії служить той чи інший вуглевод, наприклад, глюкоза. Заміна глюкози в середовищі на лактозу призводить до індукованого або адаптованого (після невеликого періоду лагфази) синтезу ферменту галактозидази (програмованого лактозним геном, див. Синтез білка), що розщеплює лактозу на глюкозу та галактозу. У тваринних тканинах подібний швидкий синтез ферментів спостерігається порівняно рідше, і механізм, що індукує синтез, вивчений тільки для невеликої кількості ферментів (тирозинтрансамінази, серин-і треоніндегідратази, триптофанпірролази та ін.). Однак при надходженні в організм деяких отрут, канцерогенних речовин, алкалоїдів, інсектицидів та ін. спостерігається різке збільшення через кілька днів активності (відповідно до кількості) ферментів - гідроксилаз гладкого ендоплазматичного ретикулуму клітин печінки; що окислюють чужорідні речовини в нетоксичні для організму продукти. З іншого боку, описані випадки, коли під дією подібних гідроксилаз чужорідні речовини перетворюються в організмі на більш токсичні сполуки. Це, зворотне детоксикації, отримало назву летального синтезу.
Проферменти.Протеолітичні ферменти шлунково-кишкового тракту та підшлункової залози синтезуються у неактивній формі, у вигляді проферментів (зимогенів). Регуляція у випадках зводиться до перетворення проферментів на активні ферменти під впливом специфічних агентів. Так, трипсин синтезується у підшлунковій залозі у формі трипсиногену. Останній перетворюється на активний трипсин у кишечнику під дією іншого білкового ферменту – ентерокінази, вперше відкритого в лабораторії І. П. Павлова. Встановлено, що активна дія ентерокінази зводиться до відщеплення від трипсиногену гексапептиду, що призводить до формування нативної третинної структури трипсину та його активного центру (див. вище); спостерігається також аутокаталіз. Перетворення неактивного пепсиногену на активний пепсин здійснюється аутокаталітично внаслідок обмеженого протеолізу в присутності НС1 і також пов'язане з відщепленням від першого специфічного інгібітора поліпептидної природи (див. Обмін простих білків). Синтез протеїназ у неактивній формі та інших неактивних білків-попередників має, очевидно, певний біологічний сенс, запобігаючи руйнуванню клітин органів, у яких утворюються проферменти.
Хімічна модифікація ферменту.Вище було зазначено (див. Хімія білків), що ряд білків при формуванні третинної структури піддається постсинтетичній модифікації. Виявилося, що ключові ферменти енергетичного обміну - фосфорилаза, глікогенсинтетаза та ін - також контролюються шляхом фосфорилювання та дефосфорилювання, що здійснюється специфічними ферментами - протеїнкіназою та протеінфосфатазою, рівень активності яких у свою чергу регулюється гормонами (див. Обмін вуглеводів). Рівень активності ключових ферментів і відповідно інтенсивність процесів обміну визначатиметься співвідношенням фосфорильованих та дефосфорильованих форм цих ферментів.
Регуляція активності ферментів за принципом зворотний зв'язок.У багатьох строго біосинтетичних реакціях основним типом регуляції швидкості багатоступінчастого ферментативного процесу є пригнічення за принципом зворотного зв'язку, коли кінцевий продукт біосинтетичного ланцюга пригнічує активність ферменту, що каталізує першу стадію.
Припустимо, що у клітинах здійснюється багатоступінчастий біосинтетичний процес, кожна стадія якого каталізується власним ферментом:

Швидкість подібної сумарної послідовності реакцій значною мірою визначається концентрацією кінцевого продукту (Р), накопичення якого вище за допустимий рівень має потужну інгібуючу дію на першу стадію процесу, відповідно на фермент E 1 .
Вперше існування такого механізму контролю активності ферментів метаболітами було показано у Е. coli при дослідженні синтезу ізолейцину та цитидинтрифосфату (ЦТФ). Виявилося, що ізолейцин, що є кінцевим продуктом, вибірково пригнічує активність треоніндегідратази, що каталізує першу ланку процесу перетворення треоніну на ізолейцин, що налічує п'ять ферментативних реакцій. Аналогічно ЦТФ як кінцевий продукт біосинтетичного шляху надає інгібуючу дію на перший фермент (аспартаттранскарбамоілазу), регулюючи тим самим свій власний синтез. Цей тип інгібування отримав назву інгібування за принципом зворотного зв'язку або ретроінгібування. Існування його доведено у всіх живих організмах, і в даний час він розглядається як один із провідних типів регуляції активності ферментів та клітинного метаболізу в цілому 1 . (1 Слід зазначити, що швидкість реакції (як і активність ферментів) у суто біодеградативних (катаболічних) процесах регулюється проміжними продуктами, що є індикаторами енергетичного стану клітини (пуринові нуклеотиди, пірофосфат, неорганічний фосфат та ін.).)
З іншого боку, в амфіболічних процесах (див. Введення в обмін речовин та енергії), що виконують одночасно біосинтетичні та біодеградативні функції 2 , доведено існування регуляції як за типом ретроінгібування, так і макроергами - індикаторами енергетичного стану клітини. (2 До амфіболічних процесів відносять такі шляхи, як гліколіз, глікогеноліз, цикл трикарбонових кислот, гексозомонофосфатний шлях, трансамінування амінокислот (див. Обмін речовин)). Для амфіболічних процесів унікальним типом регуляції, властивим тільки їм, є, крім того, активація попередником, коли перший метаболіт багатоступеневим шляхом активує фермент, що каталізує останню стадію. Так, доведено активуючий вплив глюкозо-6-фосфату, що є попередником глікогену, на фермент глікогенсинтетазу.
Подібні типи інгібування кінцевим продуктом та активування першим продуктом властиві алостеричним (регуляторним) ферментам (див. вище), коли ефектор, структурно відрізняючись від субстрату, зв'язується в особливому (алостеричному) центрі молекули ферменту, просторово віддаленому від активного центру. Тому прийнято розрізняти алостеричний тип регуляції, що включає як алостеричне інгібування, так і алостеричну активацію. Взаємоперетворення активного та неактивного алостеричного ферменту у спрощеній формі, а також конформаційні зміни, що спостерігаються при приєднанні субстрату та ефекторів, представлені на рис. 59.

Видно, що приєднання негативного ефектора до алостеричного центру викликає значні зміни конфігурації активного центру молекули ферменту і як наслідок втрату спорідненості ферменту до субстрату (освіта неактивного комплексу).
Алостеричні взаємодії проявляються у характері кривих залежно від початкової швидкості реакції від концентрації субстрату або ефектора, зокрема у S-образності кривих (відхилення від гіперболічної кривої Міхаеліса - Ментен). Це означає, що зв'язування однієї молекули субстрату полегшує зв'язування другої молекули у алостеричного центру, сприяючи тим самим підвищення швидкості реакції. Крім того, для регуляторних (алостеричних) ферментів характерна нелінійна залежність швидкості реакції від концентрації ферменту.
Інші типи регулювання активності ферментів.Існує ряд інших механізмів, які контролюють швидкість метаболічних процесів та активність внутрішньоклітинних ферментів. До подібних механізмів можуть бути віднесені конкуренція ферментів за загальний субстрат, виключення активності одного з нзоферментів (у множинних форм ферментів), вплив концентрацій кофакторів та їх форми (особливо іонів металів) та явище компартменталізації. Механізм компартменталізації відіграє, мабуть, важливу біологічну роль, просторово роз'єднуючи за допомогою біомембран ферменти від своїх субстратів (наприклад, лізосомальні ферменти: протеїнази, фосфатази, рибонуклеази та інші гідролітичні ферменти, від речовин, на які вони діють в цитоплазмі) або той самий час метаболічні процеси. Прикладом останніх можуть бути шляхи синтезу жирних кислот, що протікають в основному розчинної фракції цитоплазми, і шляхи розпаду жирних кислот, зосереджені в мітохондріях.
Визначення активності ферментів
Визначення кількісного вмісту ферментів у біологічних об'єктах є відомими труднощами, оскільки, за рідкісним винятком, ферменти в тканинах присутні в мізерно малих концентраціях. Тому про кількість ферментів судять за швидкістю реакції, що каталізується, в певних узгоджених умовах вимірювання. За оптимальних умов температури, pH середовища та повного насичення ферменту субстратом швидкість пропорційна концентрації ферменту. Про швидкість ферментативної реакції судять або за швидкістю втрат субстрату, або за швидкістю утворення продукту реакції.
Для вираження концентрації ферменту Комісією з ферментів Міжнародної біохімічної спілки рекомендовано стандартну одиницю (Е). За одиницю будь-якого ферменту приймається та кількість його, яка в оптимальних умовах каталізує перетворення 1 мкмоль субстрату на хвилину (мкмоль/хв). Запропоновано нове визначення міжнародної одиниці ферменту катал (кат, kat), що відповідає кількості ферменту, здатного викликати перетворення 1 моль субстрату на продукт в 1 с (1 моль/с). Відношення міжнародної одиниці (Е) до катала можна виразити так:
або 1 Е = 1 мкмоль · хв -1 = (1/60) мкмоль · з -1 = (1/60) мккат = 16,67 нкат. Таким чином, 1Е ферменту відповідає 16,67 нкат.
Рекомендовано, крім того, проведення вимірювання одиниць ферменту при 25°С, оптимумі pH та концентрації субстрату, що перевищує концентрацію насичення. У цих випадках швидкість відповідає нульовому порядку реакції щодо субстрату і залежатиме лише від концентрації ферменту.
Для вираження активності ферменту користуються визначенням питомої та молекулярної активності. Питому активність ферменту прийнято виражати числом одиниць ферментативної активності на 1 мг білка (чи числом каталів на 1 кг активного білка). Число молекул субстрату, що піддаються перетворенню однією молекулою ферменту за хвилину, прийнято називати числом оборотів, або молекулярною активністю. Так, одна молекула каталази еритроцитів здатна розщепити в 1 хв 5 · 10 6 молекул перекису водню 1 . (1 Для того щоб 1 атом неорганічного заліза, також каталізуючий розпад Н 2 O 2 розщеплював таке число молекул Н 2 O 2 , яке розщеплює каталаза в 1 с, знадобилося б більше 300 років. Цей приклад є наочним доказом однієї з основних властивостей ферментів - їх високої каталітичної активності.)
Викладач:к.б.н
Кузнєцова Катерина Ігорівна Механізми інактивації ферментів
1. Зміна первинної структури:
1.1. Розрив поліпептидного ланцюга:
Жорсткі умови (тривале кип'ятіння в HCl) –
гідроліз до окремих ам-т.
При нагріванні до 100 ° С (pH 7-8), гідроліз
пептидних зв'язків незначний.
Найбільш чутливими до
високотемпературного гідролізу є
пептидні зв'язки, утворені залишками
аспарагінової кислоти.
Протеази (бактеріальне забруднення, автоліз). Рішення:
інактивованих ферментів 1.2.Окислення функціональних груп ферменту
SH-групи цистеїну та індольні фрагменти
триптофану, при підвищеній температурі, можуть
окислюватися (сульфокси- з'єднання цистеїну
(SOH, SO2H) та утворюватися продукти
розкриття індольного кільця триптофану. Рішення:
Реактивувати за допомогою відновлюючих
агентів, зокрема низькомолекулярних тіолів
(наприклад, цистеїн або дитіотрейтол).
Викликають: Тіоли та інші відновлені
сполук сірки, наприклад, Na2SO3, Na2S2O3.
Продуктом відновлення дисульфідного зв'язку
(S-S) є:
1) тіольна форма (білок-SH)
2) змішаний дисульфід тіольної форми білка з
відновлюючим реагентом, наприклад
білок-S-SO3). 1.3. Розщеплення дисульфідних зв'язків
Лужний гідроліз цистеїну →дегідроаланін →
Завдяки нуклеофільним властивостям
взаємодіє з NH2-групами лізину та SHцистеїну →лізиноаланін та лантіонін.
Для повної деструкції всіх S-S зв'язків потрібні досить жорсткі умови (0,1-1М луг,
100 ° С).
Однак деструкція найбільш реакційних
S–S зв'язків може проходити у досить м'яких
умовах – наприклад, при температурах 60–80 °С та
слаболужних значеннях рН.
Слід враховувати при використанні ферментів у
як добавок до миючих засобів. Рішення:
Додавання в середу тіолів призведе до
розщепленню змішаного дисульфіду та
подальшому утворенню правильного S-S зв'язку 1.4. Хімічна модифікація каталітичних SHгруп.
Катіони важких металів (Hg, Pb та Cu)
зв'язуються з SH-груп активного центру
ферменту
↓
Утворення відповідних меркаптидів
↓
Фермент інактивується
10.
1.5. Фосфорилювання білків in vivo.Під дією фосфорилази та фосфатази,
містяться в напівочищених ферментативних
препаратах у вигляді домішок
↓
Фосфорна кислота зв'язується з ОН-групами
серина та треоніну.
↓
Конформаційні зміни в білковій
молекулі
↓
інактивацію ферменту.
11.
Рішення:У літературі практично відсутні приклади
вдалої реактивації таким чином
інактивованих ферментів
12.
1.6. Дезамінування залишків аспарагіну.При температурах (порядку 100 ° С) та рН (порядку
4,0-5,0) відбувається дезамінування залишків
аспарагіну.
↓
інактивацію ферменту.
13.
Рішення:У літературі практично відсутні приклади
вдалої реактивації таким чином
інактивованих ферментів
14.
1.7. Радіаційна інактивація ферментівγ-опромінення та УФ-світло
Впливають на функціональні групи
ферментів, пептидні зв'язки та SH-групи
залишків цистеїну.
15.
2. АгрегаціяСпостерігається при підвищеній температурі, при
екстремальних значеннях рН, у присутності
деяких хімічних сполук.
Чим вища концентрація, тим швидше йде
агрегація.
Гідрофобні взаємодії та водневі
зв'язку, можливе утворення дисульфідних
містків між окремими білковими
молекулами
16.
Рішення:Необхідно зруйнувати міжмолекулярні
ковалентні та нековалентні контакти c
допомогою концентрованих розчинів
сечовини та гуанідинхлориду, екстремальних
значень рН.
Якщо під час агрегації ферментів утворилися
міжмолекулярні S-S-містки, що в середу вносять у
щодо невисоких концентрацій
(мкмоль/л) тіоловмісні реагенти (наприклад,
цистеїн або дитіотрейтол).
При таких концентраціях внутрішньомолекулярні
S-S зв'язки в білку, як правило, не торкаються.
17.
3. Інактивація ферментів поверхневимнатягом
Поверхневий натяг на межі розділу
між повітрям та чистою водою становить 80
дін/див.
Піноутворення викликає денатурацію
ферментів, адсорбованих на межі розділу
фаз.
18.
Рішення:Додавання ПАР знижує поверхневе
натяг до 1 дин/див.
19.
4. Сорбція білка на реакційних стінкахсудини
Сорбція за рахунок нековалентних взаємодій
призводить до зменшення концентрації ферменту в
розчині.
Необхідно враховувати під час роботи з
розведеними білковими розчинами
(Концентрація 10-8-10-10 моль / л).
Під впливом денатуруючих факторів
здатність білків сорбуватися на стінках
реакційної судини може зростати.
20.
Рішення:Десорбція ферменту зі стінок реакційного
судини досягаються за рахунок руйнування
неспецифічних взаємодій між
білком та сорбційними центрами на поверхні
судини.
Можна використовувати екстремальні значення рН,
концентровані розчини сечовини або
гуанідинхлориду.
21.
5. Дисоціація олігомерних білків насубодиниці
Викликають: Сечовина, детергенти, кислоти або
нагрівання.
Приводять до:
конформаційним змінам окремих
субодиниць;
агрегації субодиниць;
дисоціації кофакторів із активних центрів;
модифікації функціональних груп, які в
олігомерному білку були екрановані від
контакту із розчинником.
22.
6. Десорбція кофактора з активного центруферменту
Викликає: нагрівання, дія хелаторів, діаліз
Якщо дисоціація кофактора супроводжується
значними конформаційними зрушеннями або
хімічною модифікацією важливих
функціональних груп → фермент
інактивується необоротно.
Якщо при цьому не сталося суттєвого
зміни білкової конформації, то додавання
у середу надлишку кофактора призводить до
реактивації ферменту.
23.
Регенерація кофакторівСпособи регенерації:
Ферментативний (методи з використанням сполучених субстратів чи ферментів)
Неферментативний (хімічні та
електрохімічні підходи)
24.
Ферментативний спосібУ систему вводять надмірну кількість
сполученого субстрату того ж ферменту:
Приклад: під час роботи алкогольдегідрогенази
витрачається НАДН.
25.
Ферментативний спосіб1. Використання сполучених субстратів.
Недоліки:
використовуються високі концентрації
сполученого субстрату, оскільки рівновага
реакції сильно зрушено убік
утворення спирту;
ускладнює процедуру виділення основного
продукту із реакційної суміші.
26.
Ферментативний спосіб2. Використання сполучених
ферментативних реакцій
В систему, додатково вводять фермент 2,
функціонування якого забезпечує
регенерацію коферменту.
Використовувані у системі ферменти повинні мати
різну субстратну специфічність
27.
Неферментативні методи1. Хімічні способи.
Використовуються дитіоніт натрію та деякі
солі піридинія:
+ Низька вартість.
- можуть пригнічувати окремі ферменти.
Флавінові коферменти
28.
Неферментативні методи2. Електрохімічні методи.
Пряме електрохімічне відновлення або
окиснення.
“-” появи у процесі регенерації
ферментативно неактивних форм коферменту,
наприклад, в результаті його димеризації.
29. СТАБІЛІЗАЦІЯ ФЕРМЕНТІВ У БІОТЕХНОЛОГІЧНИХ СИСТЕМАХ
30.
Проблеми, що виникають при використанніферментів у біотехнологічних процесах:
1. Підвищені температури
2. Екстремальні значення pH
3. Високі концентрації органічних
розчинників або ПАР.
4. Неможливість багаторазово використовувати
фермент.
5. Складність при розподілі ферменту від
продукту.
31.
Основні підходи для стабілізаціїферментів:
1. Додавання стабілізуючих речовин у середу,
в якій зберігається фермент або проводиться
ферментативна реакція
2. Хімічна модифікація ферменту.
3. Іммобілізація ферменту.
32.
1. Субстратів або їх аналогів:
Фермент-субстратний комплекс часто більше
стійкіший, ніж вільний фермент.
Приклад: Лактатдегідрогеназа у присутності
лактату більш термостійкий.
33.
Стабілізація ферментів за допомогою:2. Органічні розчинники:
Багатоатомні спирти стабілізують деякі
ферменти за рахунок підвищення стійкості
внутрішньомолекулярних водневих зв'язків білка
Приклад: Хімотрипсин у присутності 50-90%
гліцерину більш стійкий до протеолізу
34.
Стабілізація ферментів за допомогою:3. Солей:
При низьких концентраціях солей (<0,1M) катионы
Са2+, Zn2+, Mn2+, Fe2+ та ін. можуть специфічно
взаємодіяти з металопротеїнами.
Деякі з них – кофактори.
Са2+ здатний стабілізувати третинну
структуру ряду білків завдяки освіті
іонних зв'язків з двома різними
амінокислотними залишками.
Приклад: у α-амілази (з bacillus caldolyticus)
Са2+ значно підвищує термічну
стійкість.
35.
36.
Хімічна модифікація ферменту1. Фермент приймає більш стабільну
конформацію.
2. Введення в білок нових функціональних груп
призводить до утворення додаткових
стабілізуючих водневих зв'язків або сольових
містків.
3. При використанні неполярних з'єднань
посилюються гідрофобні взаємодії.
4. Модифікація гідрофобних областей поверхні
білка гідрофільними сполуками зменшує
площа несприятливого контакту зовнішніх
неполярних залишків із водою.
Приклад: глутаровий альдегід
37.
Іммобілізація ферменту дозволяє:Підвищити стійкість ферменту (нагрівання,
автолізу, дії агресивних середовищ тощо)
1. Багаторазово використовувати фермент
2. Відокремлювати фермент від реагентів та продуктів
реакції.
3. Переривати реакцію у потрібний момент.
38.
Іммобілізовані ферменти – це препаратиферментів, молекули яких пов'язані з
носієм, зберігаючи при цьому повністю або
частково свої каталітичні властивості.
Методи іммобілізації:
1. Хімічні
2. Фізичні
39.
Методи іммобілізації:Як носії можуть застосовуватися:
1) Органічні матеріали:
1.1) природні (полісахариди, білки, ліпіди)
1.2) синтетичні полімерні носії
2) Неорганічні матеріали (матриці на
основі силікагелю, глини, кераміки, природних
мінералів і т. д.)
40.
1) адсорбція ферменту на нерозчинному носії
в результаті електростатичних, гідрофобних,
вандер-ваальсових та ін. взаємодій;
;
структури;
4) Включення до двофазної системи.
41.
Методи фізичної іммобілізації:Досягається шляхом контакту водного розчину
ферменту з носієм.
42.
Методи фізичної іммобілізації:1) адсорбція ферменту на нерозчинному носії
Чинники, що впливають на адсорбцію:
1. Питома поверхня та пористість носія
2. Значення рН (на не іонообмінниках мах адсорбція
в ізоелектричній точці білка)
3. Іонна сила розчину (зростання іонної сили –
десорбція ферменту, але іноді зворотна ситуація
"висолення")
4. Концентрація ферменту.
5. Температура (з одного боку денатурація, з
інший прискорена дифузія)
43.
Методи фізичної іммобілізації:1) адсорбція ферменту на нерозчинному носії
Переваги:
2) Доступність носіїв
Недоліки:
1) Недостатня міцність зв'язування
2) Багато носіїв біодеградовані
44.
Методи фізичної іммобілізації:2) включення ферменту в напівпроникну
капсулу, напівпроникну мембрану
45.
Методи фізичної іммобілізації:2) включення ферменту в напівпроникну
капсулу, напівпроникну мембрану
Переваги:
1) Відносна простота методики
2) Захист від мікроорганізмів
3) Немає дифузійних обмежень (т.к.
відношення поверхні до площі велике та
товщина мембрани невелика)
Недоліки:
1) Біодеградовані
2) Не застосовується для високомолекулярних
46.
Методи фізичної іммобілізації:3) механічне включення ферменту в гелеві
структури
Фермент входить у тривимірну сітку
полімерних ланцюгів, що утворюють гель.
47.
Методи фізичної іммобілізації:3) механічне включення ферменту в гелеві
структури
Необхідно враховувати:
1. Відповідність розміру доби розміру ферменту.
2. Природа матриці (бо вона створює
мікрооточення для ферменту, може
створювати рН відмінне від рН розчину та
підвищувати спорідненість субстрату до матриці, що
підвищує швидкість ферментативної реакції)
48.
Методи фізичної іммобілізації:3) механічне включення ферменту в гелеві
структури
Переваги:
1) Відносна простота методики
2) Підвищена механічна, хімічна та
теплова стійкість матриць.
3) Відбувається стабілізація ферменту
4) Фермент захищений від бактеріального
ушкодження
Недоліки:
1) Не застосовується для високомолекулярних
49.
Методи фізичної іммобілізації:4) Включення до двофазної системи
Фермент розчинний тільки в одній із фаз, а
продукт - в інший
Дозволяє працювати у високомолекулярних
субстратами.
50.
Методи хімічної іммобілізації:Утворення ковалентних зв'язків між
ферментом та носієм.
Переваги:
1) Висока міцність кон'югату
2) Можна підвищувати стабільність ферменту
51.
При іммобілізації ферментів необхіднодотримуватись таких умов:
1. Активні групи матриці не повинні
блокувати каталітичний центр ферменту.
2. Іммобілізації не повинні призводити до втрати
активність ферменту.
52.
Дуже перспективним є використання вякості біокаталізаторів іммобілізованих
клітин.
Т.к. можна уникнути:
1) дорогі стадії виділення та очищення
ферментів
2) необхідності їхньої подальшої стабілізації
53.
ТермозимиСтабільні в умовах високої температури,
високих концентрацій солей та екстремальних
значень рН.
Гіпертермофільні мікроорганізми,
що зустрічаються серед Archaea і Bacteria, живуть
при температурі 80–100 °С.
54.
Механізми відповідальні за термостійкістьферментів у термозимів:
Між мезофільними та термофільними
версіями ферментів - високий рівень гомології
послідовності та структури.
Так, послідовності термостабільних
дегідрогеназ з Pyrococcus і Thermotoga на 35 і
55% відповідно ідентичні
послідовності мезофільної дегідрогенази
з Clostridium.
55.
Було виявлено, що дегідрогеназа з Pyrococcusfuriosus (Tm == 105 °C) містить 35 ізолейцинів,
в той час як дегідрогенази з Thermotoga
maritima (Tm = 95 °C) та Clostridium symbiosum (Tm
= 55 °C) тільки 21 та 20 ізолейцинів
відповідно.
Термостабільні ферменти містять менше
гліцину: Cs дегідрогеназу містить 48 залишків
гліцину, а дегідрогенази з Tm і Pf тільки
39 та 34 гліцину відповідно.
Більше ізолейцину та менше гліцину.
56.
Зросла термостабільність корелює:1) із збільшенням жорсткості білкової структури
за рахунок зменшення вмісту залишків
гліцину,
2) з поліпшенням гідрофобних контактів у ядрі
дегідрогенази з Pf в результаті заміни валіну
ізолейцином. (В результаті сайт-спрямованого
мутагенезу, що призводить до заміни ізолейцину
на валін термостабільність мутантів
зменшувалася).
57.
Механізми стабілізації:мінімізація доступної площі гідрофобної
поверхні білка;
оптимізація упаковки атомів білкової
молекули (мінімізація відношення
поверхня/обсяг);
оптимізація розподілу зарядів (досягається
завдяки усуненню відштовхувальних
взаємодій, а також у результаті організації
взаємодій між зарядами у своєрідну
мережа)
Зменшення кількості западин
58.
Застосування ферментів з екстремофілівСучасні технології молекулярної біології
та генної інженерії дозволяє:
1) отримувати достатню кількість ферментів з
екстремофілів для їх подальшого
аналізу та практичного застосування.
2) клонування та експресія цих ферментів у
мезофільні організми.
59.
Крохмаль використовується для виробництва цукрів.
Спочатку процес ведеться при (95-105 ° С) і при значеннях
рН 6-6,5.
На наступному етапі температура знижується до 60°С
рН = 4,5.
Використання термостабільних ферментів (αамілази, глюкоамілази, ксилозоізомерази),
виділених із гіпертермофілів, дозволить:
1) проводити процес в одну стадію і за одних і
тих самих умовах
2) відмовитися від дорогих іонообмінників
60.
Застосування ферментів з екстремофілів:Найбільш термостабільні α-амілази були
виявлені у archaea Pyrococcus woesei,
Pyrococcus furiosus, Desulfurococcus mucosus,
Пиродицизм abіsi і Staphylothermus
marinus. Гени амілази із Pyrococcus sp. були
клоновані та експресовані в E.coli та Bacillus
subtilis.
61.
Застосування ферментів з екстремофілів:Протеолітичні ферменти
Серинові лужні протеїнази широко
використовуються як добавки до миючих
засобам.
Протеїнази з екстремофілів зберігають
нативність при високих температурах,
при присутності високих концентрацій детергентів та
інших агентів, що денатурують. Pyrococcus,
Thermococcus, Staphylothermus, Desulfurococcus та
Sulfolobus. Максимальну активність ці
ферменти виявляють при температурах
від 90 до 110 °С і значення рН від 2 до 10
62.
Застосування ферментів з екстремофілів:ДНК-полімерази
Термостабільні ДНК-полімерази використовуються
у ПЛР і відіграють важливу роль у генній інженерії.
Термостабільні полімерази були виявлені у
гіпертермофілів Pyrococcus furiosus та Pyrococcus
litoralis, а також у термофілів Thermus aquaticus.